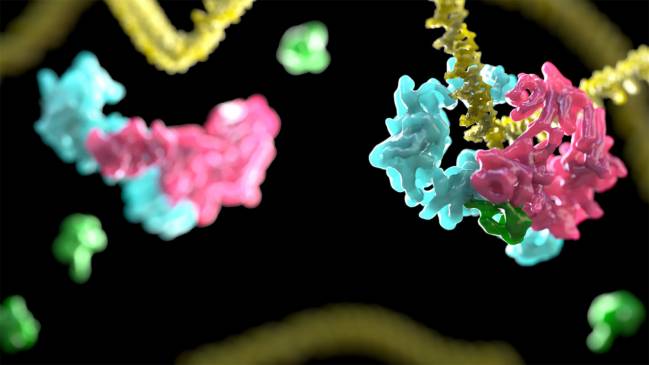
Un estudio liderado por el investigador español Pablo Alcón ha revelado cómo funciona el mecanismo molecular encargado de eliminar las lesiones en el ADN, que no funciona correctamente en los afectados por la anemia de Fanconi, una enfermedad rara hereditaria que provoca el desarrollo de cánceres, sobre todo leucemia, y fallos en la médula ósea.
La anemia de Fanconi es una enfermedad hereditaria causada por mutaciones en alguno de los genes relacionados con la reparación del ADN. Ahora, un nuevo estudio describe un nuevo gen causante de esta grave patología. Este descubrimiento es crucial no solo para el diagnóstico y el consejo genético, sino también para el desarrollo de nuevas terapias.
Un grupo de investigadores ha identificado un gen, ERCC4, que está implicado en tres enfermedades humanas según el tipo de mutación que presenta: la anemia de Fanconi, el xeroderma pigmentosum y un tipo de progeria. Los resultados, publicados hoy en la revista American Journal of Human Genetics, mejoran el conocimiento de dos rutas de reparación del ADN importantes para mantener la estabilidad de nuestros genes y prevenir el cáncer en la población general.
Un consorcio de 32 científicos de todo el mundo, liderado por Jordi Surrallés, investigador de la Universidad Autónoma de Barcelona (UAB) y miembro del Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), ha caracterizado genética y clínicamente a la práctica totalidad de pacientes españoles afectados de anemia de Fanconi, una enfermedad rara que afecta a una de cada 500.000 personas y que se caracteriza por una anemia grave a edades pediátricas, malformaciones congénitas y una alta predisposición al cáncer.