Investigadores de la Universidad de Málaga han desarrollado una aplicación bioinformática para estudiar a gran escala las relaciones entre las enfermedades genéticas y los genes que las causan. La aplicación, que se ha comenzado a utilizar en el Centro de Investigación Biomédica en Red de Enfermedades Raras, se ha publicado recientemente en la revista PLoS ONE.
Investigadores españoles han puesto en marcha una base de datos para recoger información sobre síndromes diabéticos raros, que constituyen un grupo de patologías hereditarias muy poco frecuentes cuyo nexo común es la intolerancia del organismo a la glucosa. El registro se centra en tres enfermedades minoritarias: los síndromes de Wolfram, Bardt-Biedl y Alström.
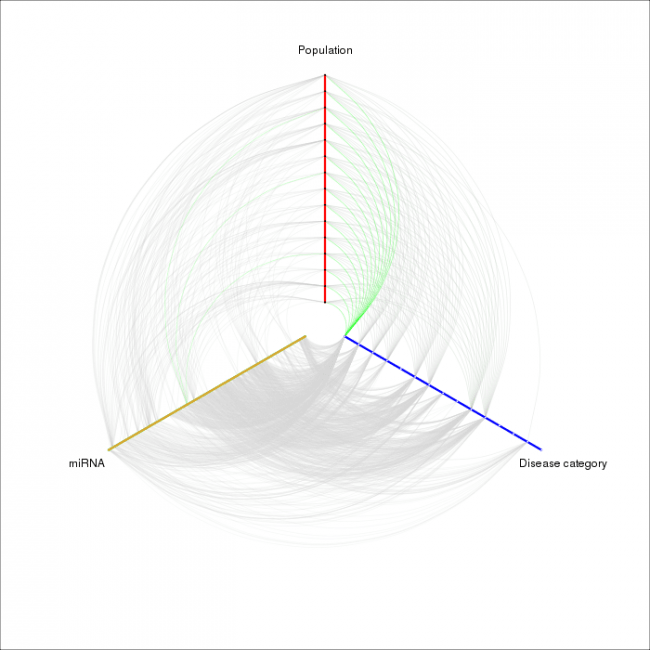
Científicos españoles han elaborado un catálogo de las variedades existentes en ciertas moléculas de ARN que son necesarias para que el mensaje de los genes sea efectivo. Hasta el momento no existía ningún trabajo sobre su variabilidad en la población general a partir de datos de secuenciación.
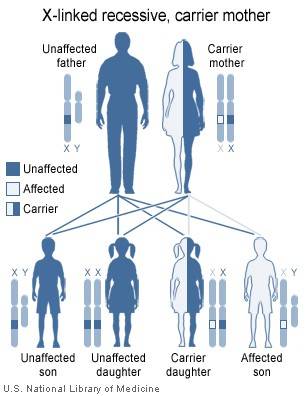
El defecto MCT8 es una extraña y grave enfermedad neurológica que se manifiesta durante los primeros meses de vida, solo en varones. Los últimos avances que han ayudado a entender y diagnosticar esta enfermedad, entre los que destaca la elaboración de una guía clínica, se publican en un reciente artículo de actualización.
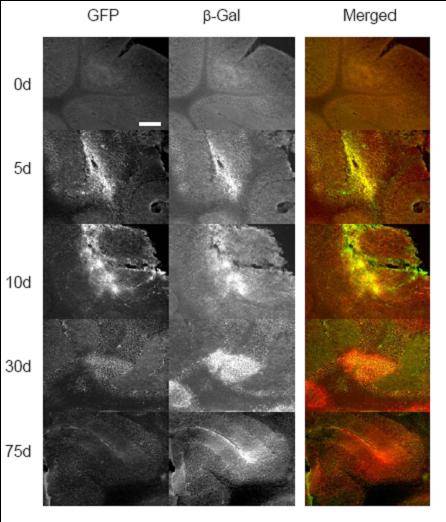
Investigadores de la Universidad Autónoma de Madrid, el CIBERER y la Universidad de Oxford han logrado comprobar que los vectores herpesvirales portadores de genes completos aseguran la persistencia de la expresión génica a largo plazo.
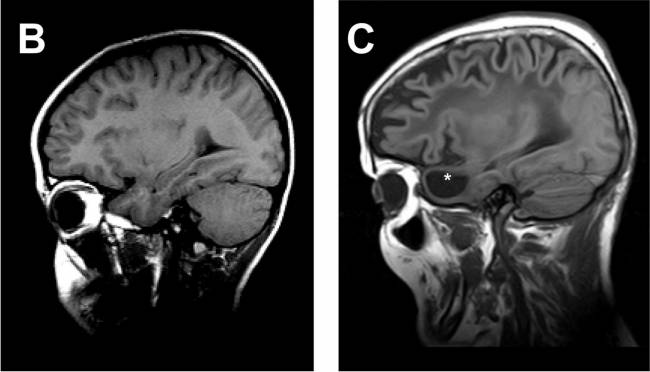
Investigadores del Instituto de Investigación Biomédica de Bellvitge (IDIBELL) y de la Universidad de Barcelona han analizado la primera muestra de un cerebro completo de una paciente afectada por leucodistrofia MLC. Este estudio ha permitido definir la relación funcional que existe entre los dos genes que causan la enfermedad, lo que abre la puerta a nuevas estrategias terapéuticas por tratar esta enfermedad rara. El estudio se ha publicado en la revista Human Molecular Genetics.
La Comisión Europea ha designado un medicamento huérfano para el tratamiento de la Anemia de Fanconi. Se trata de un vector lentiviral que contiene el gen de esta enfermedad. En la designación de este medicamento huérfano, obtenida por el Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) y el Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas (CIEMAT), han colaborado Genoma España y la Fundación Botín, entre otras instituciones.
Tras la discusión de los distintos enfoques en relación con la epidermolisis bullosa, los expertos reunidos en el Centro de Investigaciones Energéticas y Medioambientales (CIEMAT) el 2 y 3 de octubre han contrastado sus experiencias para aliviar las consecuencias de la epidermolisis bullosa (o síndrome de piel de cristal) en niñas y niños, los llamados "niños mariposa".